

Цель исследования – определить представленность повышенной трабекулярности и некомпактной кардиомиопатии в когорте больных с катехоламинергической полиморфной желудочковой тахикардией (КПЖТ).
Материал и методы. В исследование включены 68 больных с КПЖТ, наблюдающихся в Институте им. Ю. Е. Вельтищева. Для выявления наличия структурных изменений всем больным проведена эхокардиография экспертного класса, магнитно-резонансная томография сердца по показаниям.
Результаты. У детей с КПЖТ частота повышенной трабекулярности миокарда левого желудочка / некомпактной кардиомиопатии составила 24% случаев.
Заключение. У больных с КПЖТ ассоциированным структурным изменением миокарда является повышенная трабекулярность миокарда левого желудочка и некомпактная кардиомиопатия.
Предпросмотр статьи



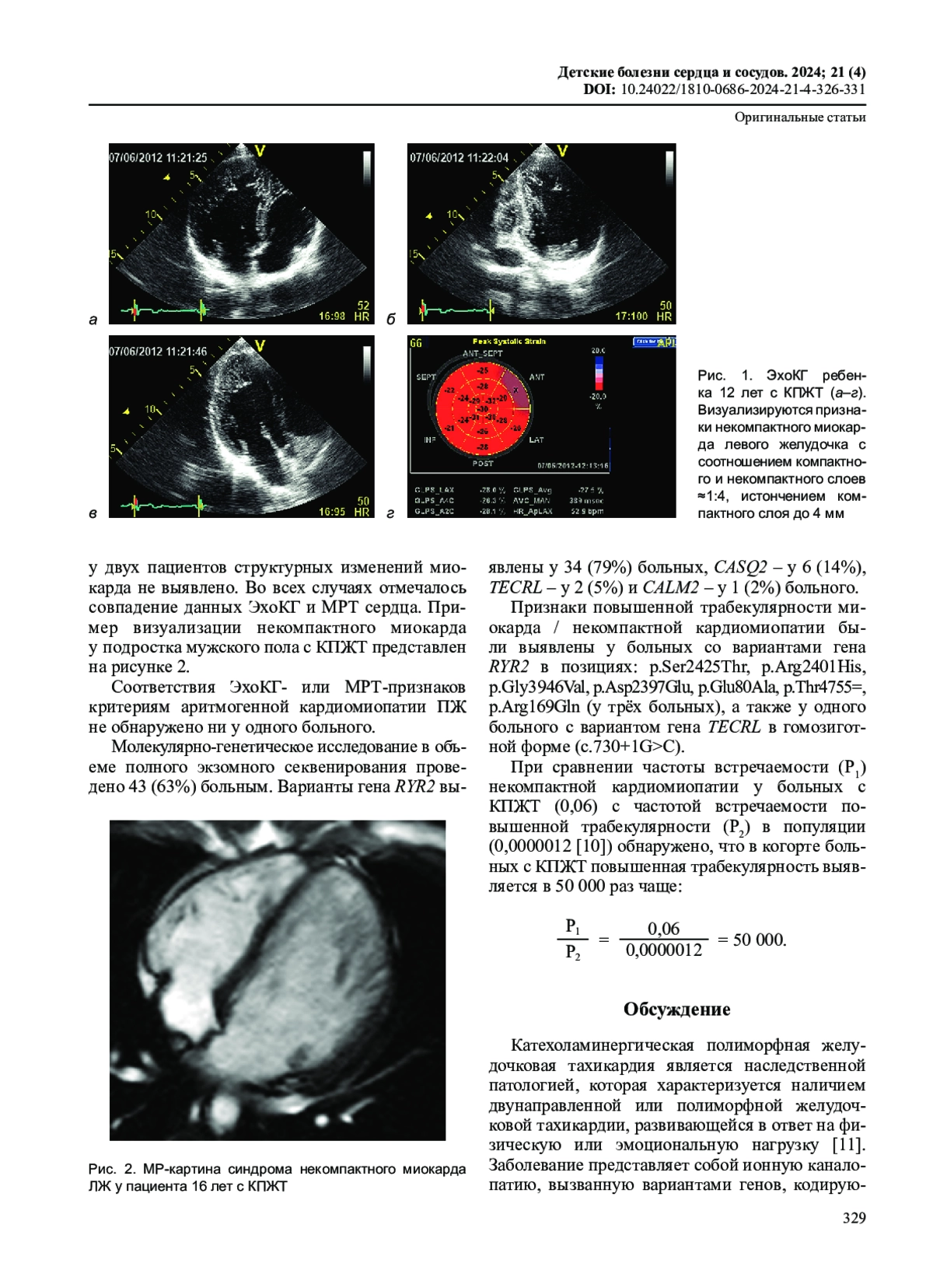


Идентификаторы и классификаторы
Согласно текущим клиническим рекомендациям, диагноз «катехоламинергическая полиморфная желудочковая тахикардия» (КПЖТ) подразумевает наличие первичной электрической патологии сердца в отсутствие ишемических и структурных нарушений, в том числе кардиомиопатий [1]. Распространенность заболевания достоверно не известна и оценивается как 1:10 000 [1]. Однако в последнее время в литературе появились публикации клинических наблюдений, свидетельствующие о возможной ассоциации повышенной трабекулярности / некомпактного миокарда и КПЖТ [2, 3].
Список литературы
1. Zeppenfeld К., Tfelt-Hansen J., de Riva M., Winkel B.G., Behr E.R., Blom N.A. et al. Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022; 43 (40): 3997–4126. DOI: 10.1161/CIR.0000000000000548
2. Roston T.M., Guo W., Krahn A.D., Wang R., Van Petegem F., Sanatani S. et al. A novel RYR2 loss-of-function mutation (I4855M) is associated with left ventricular noncompaction and atypical catecholaminergic polymorphic ventricular tachycardia. Electrocardiol. 2017; 50 (2): 227–233. DOI: 10.1016/j.jelectrocard.2016.09.006
3. Nozaki Y., Kato Y., Uike K., Yamamura K., Kikuchi M., Yasuda M. et al. Co-phenotype of left ventricular noncompaction cardiomyopathy and atypical catecholaminergic polymorphic ventricular tachycardia in association with R169Q, a ryanodine receptor type 2 missense mutation. Circ. J. 2020; 84 (2): 226–234. DOI: 10.1253/circj.CJ-19-0720
4. Chin T.K., Perloff J.K., Williams R.G., Jue K., Mohrmann R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation. 1990; 82 (2): 507–513. DOI: 10.1161/01.cir.82.2.507
5. Jenni R., Oechslin E., Schneider J., Attenhofer Jost C., Kaufmann P.A. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001; 86: 666–671. DOI: 10.1136/heart.86.6.666
6. Campbell M.J., Czosek R.J., Hinton R.B., Miller E.M. Exon 3 deletion of ryanodine receptor causes left ventricular noncompaction, worsening catecholaminergic polymorphic ventricular tachycardia, and sudden cardiac arrest. Am. J. Med. Genet. A. 2015; 167 A (9): 2197–2200. DOI: 10.1002/ ajmg.a.37140
7. Szentpali Z., Szili-Torok T., Caliskan K. Primary electrical disorder or primary cardiomyopathy? A case with a unique association of noncompaction cardiomyopathy and cathecolaminergic polymorphic ventricular tachycardia caused by ryanodine receptor mutation. Circulation. 2013; 127 (10): 1165–1166. DOI: 10.1161/CIRCULATIONAHA.112.144949
8. Petersen S.E., Selvanayagam J.B., Wiesmann F., Robson M.D., Francis J.M., Anderson R.H. et al. Left ventricular non-compaction: insights from cardiovascular magnetic resonance imaging. J. Am. Coll. Cardiol. 2005; 46 (1): 101–105. DOI: 10.1016/j.jacc.2005.03.045
9. Corrado D., Perazzolo Marra M., Zorzi A., Beffagna G., Cipriani A., Lazzari M.D. et al. Diagnosis of arrhythmogenic cardiomyopathy: the Padua criteria. Int. J. Cardiol. 2020; 319: 106–114. DOI: 10.1016/j.ijcard.2020.06.005
10. Rohde S., Muslem R., Kaya E., Dalinghaus M., van Waning J.I., Majoor-Krakauer D. et al. State-of-the art review: noncompaction cardiomyopathy in pediatric patients. Heart Fail. Rev. 2022; 27 (1): 15–28. DOI: 10.1007/s10741-021-10089-7
11. Roston T.M., Yuchi Z., Kannankeril P.J., Hathaway J., Vinocur J.M., Etheridge S.P. et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: findings from an international multicentre registry. Europace. 2018; 20 (3): 541–547. DOI: 10.1093/ europace/euw389
12. Kushnir A., Wajsberg B., Marks A.R. Ryanodine receptor dysfunction in human disorders. Biochim. Biophys Acta Mol. Cell. Res. 2018; 1865: 1687–1697. DOI: 10.1016/j. bbamcr.2018.07.011
13. Woll K.A., Van Petegem F. Calcium-release channels: structure and function of IP3 receptors and ryanodine receptors. Physiol. Rev. 2022; 102: 209–268. DOI: 10.1152/ physrev.00033.2020
14. Fowler E.D., Zissimopoulos S. Molecular, subcellular, and arrhythmogenic mechanisms in genetic RyR2 disease. Biomolecules. 2022; 12 (8): 1030. DOI: 10.3390/biom12081030
15. Ohno S., Omura M., Kawamura M., Kimura H., Itoh H., Makiyama T. et al. Exon 3 deletion of RYR2 encoding cardiac ryanodine receptor is associated with left ventricular non-compaction. Europace. 2014; 16 (11): 1646–1654. DOI: 10.1093/europace/eut382
Выпуск

Другие статьи выпуска

Синдром удлиненного интервала QT (СУИQТ) – заболевание с высоким риском развития опасных аритмий. В статье представлены результаты ЭКГ-обследования, велоэргометрии и холтеровского мониторирования пациентки 16 лет с синкопальными состояниями в анамнезе. На ЭКГ отмечено удлинение корригированного интервала QT (QTc) до 522 мс с поздним началом зубца T. Генетический тест подтвердил тип LQT3 (мутация в гене SCN5A). При холтеровском мониторировании и велоэргометрии была впервые выявлена альтернация комплекса QRS в сочетании с альтернацией зубца T (АQRS/TA). Проведен обзор литературы по проблеме.
АQRS/TA – это новый паттерн ЭКГ у пациентов с LQT3, который с высокой вероятностью отражает повышенную аритмогенную готовность миокарда у таких больных. Однако это предположение требует дальнейшего изучения и наблюдения.

Цель исследования – изучить структурно-функциональное состояние правых отделов сердца у молодых пациентов с желудочковой электрокардиостимуляцией в отдаленном послеоперационном периоде.
Материал и методы. Обследованы 58 пациентов (34 мужчины и 24 женщины) с имплантированными электрокардиостимуляторами (ЭКС) по поводу атриовентрикулярной (АВ) блокады. В зависимости от причины возникновения АВ-блокады пациентов разделили на две группы. В 1-ю группу (ЭКС+, ВПС+) вошли 28 человек с постоянным ЭКС, имплантированным после хирургической коррекции врожденного порока сердца по поводу возникшей послеоперационной АВ-блокады; во 2-ю группу (ЭКС+, ВПС-) – 30 человек с нехирургической АВ-блокадой, потребовавшей имплантации постоянного ЭКС. Всем пациентам проведены общеклиническое обследование, эхокардиография. Возраст пациентов на момент исследования составил: в 1-й группе – 21,7 (19,2; 23,3) года, во 2-й группе – 22,7 (20,1; 24,7) года; длительность электрокардиостимуляции – 15,9 (13,5; 18,2) и 15,7 (13,9; 18,5) года соответственно. Всем пациентам обеих групп на момент исследования были имплантированы двухкамерные ЭКС со 100% желудочковой стимуляцией.
Результаты. При анализе данных эхокардиографии установлено, что у пациентов 1-й группы статистически значимо большие размеры правого предсердия (ПП) по сравнению со 2-й группой: индекс объема ПП – 27,2 (23,6; 32,2) и 24,2 (21,6; 27,7) мл/м2 соответственно (U = 287,0, р = 0,039). У пациентов 1-й группы в сравнении со 2-й группой установлено значимое снижение показателей продольной функции правого желудочка (ПЖ): значения S’ составили 9,0 (8,3; 10,0) и 11,0 (11,0; 13,0) см/с соответственно (р = 0,000); TAPSE – 15,0 (13,0; 16,0) и 19,0 (18,0; 21,0) мм соответственно (р = 0,000). Показатель фракционного изменения площади (ФИП) в группах значимо не отличался и составил 51,2 (45,2; 56,2) и 46,7 (43,4; 53,2)% соответственно (U = 313,5, р = 0,098). Признаки диастолической дисфункции выявлены у 89% пациентов 1-й группы и 53% исследуемых 2-й группы (F = 0,156, p = 0,004). При оценке тяжести патологии клапанного аппарата правых отделов сердца установлено, что у пациентов 1-й группы выраженная регургитация на трикуспидальном клапане (ТК) имелась в 21%, а умеренно выраженная регургитация на клапане легочной артерии (КЛА) – в 36% случаев. У пациентов 2-й группы выраженная регургитация на ТК не регистрировалась (F = 0,124, p = 0,009) и умеренно выраженная недостаточность на КЛА выявлена у 1 пациента (F = 0,170, p = 0,002). Прогрессирование недостаточности на ТК до умеренной и выше отмечено у 78,5% исследуемых 1-й группы и 94,0% пациентов 2-й группы.
Заключение. У пациентов с послеоперационной АВ-блокадой, возникшей после хирургического лечения ВПС, в отдаленном послеоперационном периоде отмечены расширение полости ПП и снижение продольной функции ПЖ, при нормальных значениях ФИП. У пациентов с нехирургической АВ-блокадой не выявлено значимых нарушений геометрии правых отделов сердца и систолической функции ПЖ при наличии умеренной регургитации на ТК.

Цель исследования – проанализировать динамику клинических проявлений у детей с рестриктивной кардиомиопатией (РКМП), обусловленной мутациями в гене TNNI3.
Материал и методы. Проведено исследование, включающее наблюдение за 16 детьми в период с 2013 по 2024 г.
Результаты. У большинства пациентов с генетически детерминированной РКМП, обусловленной мутациями в гене TNNI3, семейный анамнез не был отягощен (94%), характер de novo установлен в 6 (37,5%) исследованных случаях (10 семей не были обследованы из-за отказа родителей). Как минимум 62,5% пациентов достигли конечной точки (летальный исход или трансплантация сердца) за период динамического наблюдения. Отмечалось более тяжелое течение заболевания у детей с дебютом в раннем возрасте на фоне крайне ограниченных возможностей медикаментозной терапии и потребностей в хирургическом лечении в короткие сроки после дебюта заболевания. В качестве иллюстрации приведен клинический пример течения заболевания у ребенка раннего возраста.
Заключение. Генетическая верификация диагноза помогает сделать прогноз заболевания, который является крайне неблагоприятным у пациентов с РКМП, обусловленной мутациями в гене TNNI3, а также решить вопрос о своевременном проведении трансплантации сердца.

Цель исследования – сравнение особенностей течения и исходов саркомерной и РАС-ассоциированной гипертрофической кардиомиопатии (ГКМП) у детей с дебютом на 1-м году жизни.
Материал и методы. По результатам генетического исследования отобраны 36 пациентов с мутациями в генах саркомерных белков и RAS-MAPK сигнального пути. В данных группах проведен сравнительный анализ клинического статуса, лабораторно-инструментальных данных, а также исходов. Разница между группами оценивалась с помощью методов биомедицинской статистики.
Результаты. У 17 (47%) пациентов наблюдались мутации в генах саркомерных белков, у 19 (53%) – в RAS-MAPK сигнального пути. Среди саркомерных мутаций в данной возрастной группе преобладали мутации в гене MYH7 (58,8%, n = 10), а в группе РАС-ассоциированной ГКМП – PTPN11 (42,11%, n = 8) и RAF1 (42,11%, n = 8). У детей с РАСопатиями наблюдалась высокая частота встречаемости врожденных пороков развития органов и систем, а также ВПС (p < 0,05). Бивентрикулярная форма ГКМП выявлена у 14 (38,9%) детей, обструктивная форма – у 17 (47,2%) пациентов. Общая выживаемость на сегодняшний момент (средняя длительность наблюдения 10 [6,0; 20,0] лет) составляет 83,3% (n = 30). Зарегистрировано 6 летальных исходов (по 3 в каждой группе). Общая выживаемость к 5 годам жизни среди всех детей составила 91,7% [95% ДИ 96,4–87,1%], к 10 годам жизни – 88,6% [95% ДИ 93,9–83,2%]. Имплантируемый кардиовертер-дефибриллятор был установлен 4 (23,5%) детям только с саркомерной ГКМП. Миоэктомия проведена 9 (25%) пациентам.
Заключение. Течение ГКМП у детей с дебютом до года характеризуется высокой летальностью. Дети с РАСопатиями представляют особую группу риска в раннем возрасте.

Цель исследования – оценить распространенность тахииндуцированной кардиомиопатии (ТКМП) и выявить факторы, ассоциированные с риском развития ТКМП у детей с идиопатическими желудочковыми аритмиями (ЖА).
Материал и методы. В ретроспективное исследование включены 392 ребенка с идиопатическими ЖА, которым было проведено лечение в НМИЦ им. В. А. Алмазова в 2011–2023 гг. В 1-ю группу вошли 35 (8,9%) пациентов с наличием ТКМП, в группу 2 – 357 (91,1%) пациентов без критериев ТКМП. Всем пациентам с ТКМП проводилась оценка эхокардиографических данных через 2 и 6 мес после старта терапии или проведения катетерной аблации.
Результаты. Распространенность ТКПМ у детей с идиопатическими ЖА составила 8,9%. В ходе исследования были выявлены статистически значимые факторы риска формирования ТКМП у детей с ЖА: возраст 12 лет и старше (p = 0,008), наличие жалоб на повышенную утомляемость (p = 0,040) и пресинкопальные/синкопальные состояния (p = 0,035), наличие парных желудочковых экстрасистол (ЖЭ) (p = 0,010) и желудочковой тахикардии (ЖТ) (p = 0,002). Методом ROC-анализа установлено, что суточная плотность желудочковой эктопии 36,8% и более ассоциируется с развитием ТКМП (p < 0,001). Чувствительность и специфичность модели составили 76,5 и 78,1% соответственно.
Заключение. Такие факторы, как возраст 12 лет и старше, наличие симптомов, парных ЖЭ, ЖТ, а также плотность ЖА, могут использоваться в клинической практике в качестве предикторов формирования ТКМП у детей с идиопатическими ЖА.

Цель исследования – изучить влияние мутаций генов, ответственных за развитие семейной гиперхолестеринемии (СГХС), и липидного профиля на структурно-морфологическое состояние сосудов по показателям толщины комплекса интима–медиа (тКИМ) общей сонной артерии у детей с гетерозиготной семейной гиперхолестеринемией.
Материал и методы. Исследование проводилось в период с 2019 по 2023 г. и включало 214 детей в возрасте от 2 до 17 лет. Группу сравнения составили 107 детей. В основную группу вошли 107 пациентов с диагнозом «семейная гиперхолестеринемия, гетерозиготная форма», которые были разделены на три группы в зависимости от генофенотипа заболевания: группа 2 – дети с положительным фенотипом и с идентифицированными мутациями, ассоциированными с СГХС: LDLR, APOB, PCSK9, LDLRAP1; группа 3 – дети с положительным фенотипом, но не выявленными мутациями; группа 4 – дети с отрицательным фенотипом и положительным генотипом. Всем детям проводились клинико-лабораторная диагностика, УЗИ сосудов шеи с оценкой тКИМ общей сонной артерии. Пациентам основной группы было проведено секвенирование нового поколения с применением панели генов: LDLR, APOB, PCSK9, LDLRAP1.
Результаты. У пациентов 2-й и 3-й групп выявлено статистически значимое увеличение показателей общего холестерина (ОХ), липопротеинов низкой плотности (ЛПНП) и тКИМ относительно группы сравнения. Значения тКИМ были достоверно больше у детей 2-й группы относительно 3-й, при этом показатели липидов у них не отличались. В 4-й группе показатели ЛПНП и тКИМ были достоверно ниже аналогичных показателей 2-й группы. Наиболее распространенными вариантами LDLR оказались: c.986G>A, c.906C>G, c.1187-10G>A. Увеличение тКИМ было достоверно выше у пациентов с c.1187-10G>A относительно c.906C>G.
Заключение. Пациенты с СГХС характеризовались увеличенными уровнями ОХ, ЛПНП и большей тКИМ в отличие от здоровых сверстников. У детей с идентифицированной мутацией в генах, ассоциированных с СГХС, установлены признаки сосудистого ремоделирования с 8-летнего возраста по сравнению с детьми с фенотипической СГХС. Полученные результаты подчеркивают важность ранней диагностики, в том числе генетической, и постоянного мониторинга состояния сосудистой стенки пациентов с СГХС для предотвращения прогрессирования атеросклероза начиная с детского возраста.

Мышечная дистрофия Дюшенна (МДД) в настоящее время представляет собой одну из самых изучаемых прогрессирующих мышечных дистрофий, в отношении которой ведется интенсивная разработка новых фармакологических методов лечения, направленных на коррекцию генетического дефекта и восстановление экспрессии дистрофина. Самыми многообещающими и наиболее перспективными подходами для терапии являются пропуск экзона и технология первичного редактирования с помощью CRISPR/Cas9. Однако, несмотря на эффективность новых методик лечения, большую роль в определении прогноза и продолжительности жизни больных с МДД продолжает играть неуклонно развивающаяся кардиомиопатия, приводящая к декомпенсированной сердечной недостаточности и жизнеугрожающим нарушениям ритма. Прогрессирование кардиомиопатии влияет на доступность всех новых методов лечения, снижая их эффективность и общую функциональную пользу. Поэтому в лечении МДД важен комбинированный подход, включающий в себя как генную терапию, направленную на восстановление экспрессии дистрофина, так и терапию, ориентированную на улучшение выживаемости структурно неполноценных кардиомиоцитов, уменьшение выраженности фиброза миокарда и смягчение дистрофических процессов. В данном обзоре приводится современный взгляд на концепцию единства медикаментозных, хирургических и генотерапевтических методов лечения МДД, целью которых является уменьшение выраженности миокардиальной дисфункции.
Статистика статьи
Статистика просмотров за 2025 год.
Издательство
- Издательство
- ФГБУ НМИЦ ССХ ИМ. А.Н. БАКУЛЕВА МИНЗДРАВА РОССИИ
- Регион
- Россия, Москва
- Почтовый адрес
- 121552, Москва, Рублевское шоссе, д. 135.
- Юр. адрес
- 119049, г Москва, р-н Якиманка, Ленинский пр-кт, д 8
- ФИО
- Голухова Елена Зеликовна (ДИРЕКТОР)
- Контактный телефон
- +7 (495) 4147984
- Сайт
- https://bakulev.ru/