

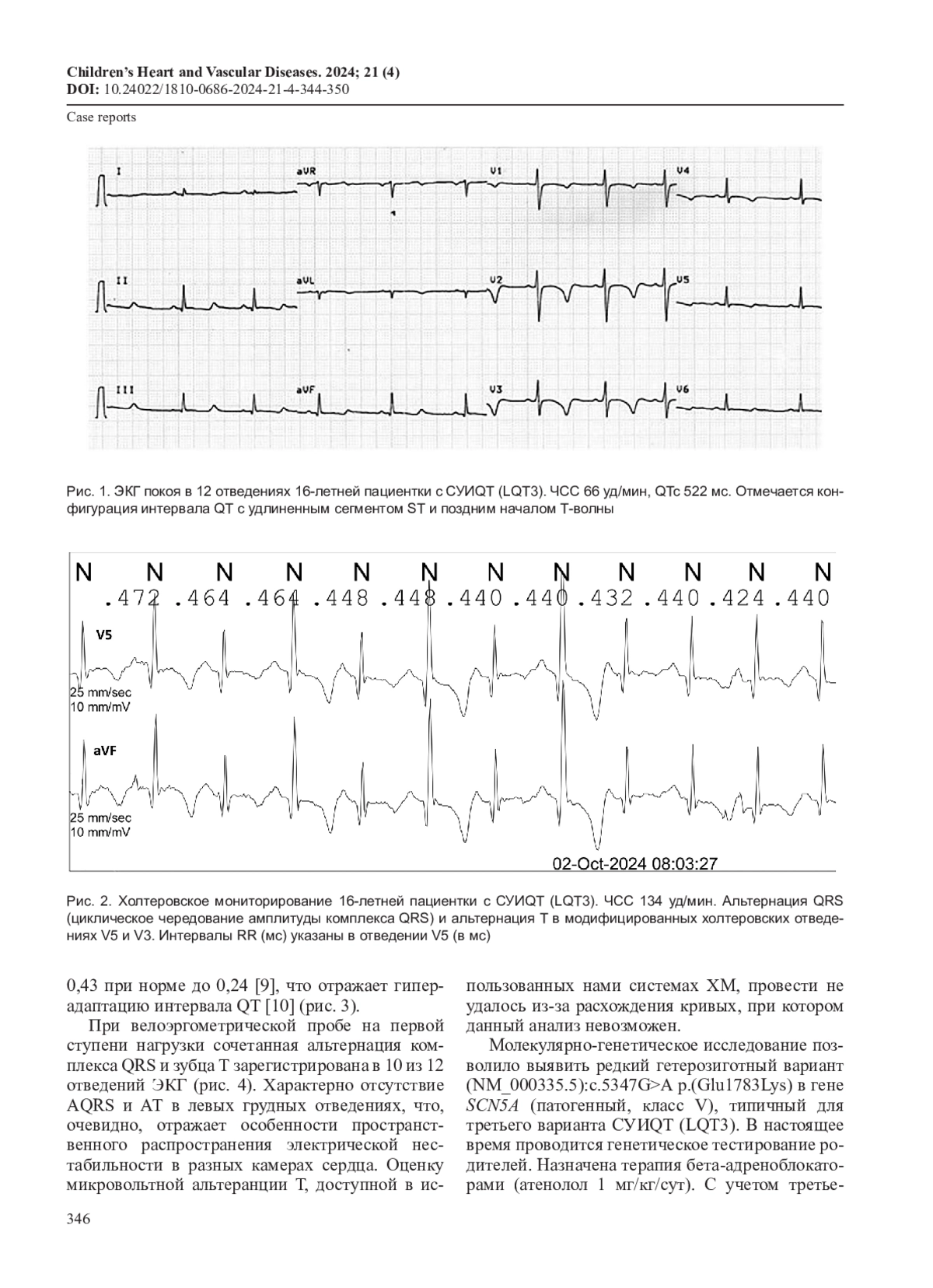
Синдром удлиненного интервала QT (СУИQТ) – заболевание с высоким риском развития опасных аритмий. В статье представлены результаты ЭКГ-обследования, велоэргометрии и холтеровского мониторирования пациентки 16 лет с синкопальными состояниями в анамнезе. На ЭКГ отмечено удлинение корригированного интервала QT (QTc) до 522 мс с поздним началом зубца T. Генетический тест подтвердил тип LQT3 (мутация в гене SCN5A). При холтеровском мониторировании и велоэргометрии была впервые выявлена альтернация комплекса QRS в сочетании с альтернацией зубца T (АQRS/TA). Проведен обзор литературы по проблеме.
АQRS/TA – это новый паттерн ЭКГ у пациентов с LQT3, который с высокой вероятностью отражает повышенную аритмогенную готовность миокарда у таких больных. Однако это предположение требует дальнейшего изучения и наблюдения.
Предпросмотр статьи





Идентификаторы и классификаторы
Синдром удлиненного интервала QT (СУИQТ) – генетически детерминированное заболевание с высоким риском развития аритмогенных жизнеугрожающих состояний – полиморфной желудочковой тахикардии torsade de pointes (TdP), синкопе, остановки сердца и внезапной смерти. Данные состояния могут быть первым и единственным клиническим проявлением заболевания, поэтому крайне важно искать ранние, доклинические маркеры риска их развития. Некоторые изменения ЭКГ, такие как продолжительность интервала QT, желудочковые аритмии и альтернация зубца T, отнесены к прогностически неблагоприятным как при СУИQT [1], так и при других заболеваниях с риском внезапной смерти [2].
Список литературы
1. Wilde A.A.M., Amin A.S., Postema P.G. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart. 2022; 108 (5): 332–338. DOI: 10.1136/ heartjnl-2020-318259
2. Calò L., Lanza O., Crescenzi C., Parisi C., Panattoni G., Martino A. et al. The value of the 12-lead electrocardiogram in the prediction of sudden cardiac death. Eur. Heart J. 2023; 25 (Suppl. C): C218–C226. DOI: 10.1093/ eurheartjsupp/suad023
3. Makarov L., Akopyan A., Komoliatova V. The ECG of a 6-year-old girl. J. Electrocardiol. 2024; 87: 153819. DOI: 10.1016/j.jelectrocard.2024.153819
4. Макаров Л.М., Акопян А.Г., Комолятова В.Н., Беспорточный Д.А. Сочетанная альтернация комплекса QRS и зубца Т – новый электрокардиографический феномен при синдроме удлиненного интервала QT: клиническое наблюдение. Российский кардиологический журнал. 2024; 29 (10S): 6036. DOI: 10.15829/1560-4071- 2024-6036 Makarov L.M., Akopyan A.G., Komolyatova V.N., Besportochny D.A. Combined QRS complex and T wave alternans – a new electrocardiographic phenomenon in long QT syndrome: a clinical observation. Russian Journal of Cardiology. 2024; 29 (10S): 6036 (in Russ.). DOI: 10.15829/ 1560-4071-2024-6036
5. Marquez M.F., Allende R., Morales J.L. Unmasking the Brugada syndrome with high parasternal leads. Europace. 2007; 9 (12): 1216. DOI: 10.1093/europace/eum229
6. Zareba W., Moss A.J., le Cessie S., Hall W.J. T wave alternans in idiopathic long QT syndrome. J. Am. Coll. Cardiol. 1994; 23 (7): 1541–1546. DOI: 10.1016/0735- 1097(94)90653-x
7. Сигал А.М. Ритмы сердечной деятельности и их нарушения. Одесса: Одобллит; 1935. Seagal A.M. Rhythms of cardiac activity and their disorders. Odessa; 1935 (in Russ).
8. Makarov L., Komoliatova V., Zaklyazminskaya E., Dmitrieva A.V. Ambulatory E.C.G. monitoring in patients with long QT syndrome. Conference: Scientific Session AHA.Su 3177. Chicago, November 10–12, 2018. Chicago; 2018. DOI: 10.13140/RG.2.2.32275.69921
9. Макаров Л.М., Комолятова В.Н., Мирошникова Е.Н., Казанцева МА. Физиологическое значение и нормативные параметры частотной адаптации QT интервала при холтеровском мониторировании у здоровых молодых лиц. Кардиология. 2008; 48 (4): 54–58. Makarov L.M., Komoliatova V.N., Miroshnikova E.N., Kazantseva M.A. Physiological significance and normative parameters of rate adaptation of QT-interval during holter monitoring in healthy persons of young age. Kardiologiia. 2008; 48 (4): 54–58 (in Russ.).
10. Макаров Л.М. Звено патогенеза ночной внезапной смерти при сердечной недостаточности. Медицина экстремальных ситуаций. 2022; 24 (3): 74–76. DOI: 10.47183/ mes.2022.026 Makarov L.М. The component of pathogenesis of sudden nocturnal death in patients with heart failure. ExtremeMedicine. 2022; 24 (3): 74–76 (in Russ.). DOI: 10.47183/ mes.2022.026
11. Schwartz P.J., Crotti L., Insolia R. Long-QT syndrome: from genetics to management. Circ. Arrhythm. Electrophysiol. 2012; 5 (4): 868–877. DOI: 10.1161/CIRCEP. 111.962019
12. Chorin E., Taub R., Medina A., Flint N., Viskin S., Benhorin J. Long-term flecainide therapy in type 3 long QT syndrome. Europace. 2018; 20 (2): 370–376. DOI: 10.1093/ europace/euw439. PMID: 28339995
13. Макаров Л.М., Комолятова В.Н., Киселева И.И., Акопян А.Г. Синдром удлиненного интервала QT. В кн.: Макаров Л.М., Комолятова В.Н. (ред.). Внезапная сердечная смерть у детей, подростков и молодых лиц. М.: Медпрактика; 2021: 222–246. Makarov L.M., Komolyatova V.N., Kiseleva I.I., Akopyan A.G. Long QT syndrome. In: Makarov L.M., Komolyatova V.N. (Eds). Sudden cardiac death for children, adolescents and young adults. Moscow; 2021: 222–246 (in Russ.).
14. Horne A.J., Eldstrom J., Sanatani S., Fedida D. A novel mechanism for LQT3 with 2:1 block: a pore-lining mutation in Nav1.5 significantly affects voltage-dependence of activation. Heart Rhythm. 2011; 8 (5): 770–777. DOI: 10.1016/ j.hrthm.2010.12.041
15. Макаров Л.М., Акопян А.Г., Заклязьминская Е.В., Комолятова В.Н., Исланов И.О., Беспорточный Д.А. Новый электрокардиографический феномен при синдроме удлиненного интервала QT: клиническое наблюдение. Педиатрия им. Г.Н. Сперанского. 2024; 103 (5): 126–131. DOI: 10.24110/0031-403X-2024-103-5-126-131 Makarov L.M., Akopyan A.G., Zaklyazminskaya E.V., Komolyatova
V.N., Islanov I.O., Besportochnyj D.A. Clinical case of a newly spotted electrocardiographic phenomenon in long QT syndrome. Pediatria n.a. G.N. Speransky. 2024; 103 (5): 126–131 (in Russ.). DOI: 10.24110/0031-403X- 2024-103-5-126-131
16. Makarov L., Komoliatova V. Microvolt T-wave alternans during Holter monitoring in children and adolescents. Ann. Noninv. Electrocardiol. 2010; 15 (2): 138–144. DOI: 10.1111/ j.1542-474X.2010.00354.x
17. Takasugi N., Goto H., Takasugi M., Verrier R.L., Kuwahara T., Kubota T. et al. Prevalence of microvolt T-wave alternans in patients with long QT syndrome and its association with torsade de pointes. Circ. Arrhythm. Electrophysiol. 2016; 9 (2): e003206. DOI: 10.1161/CIRCEP.115.003206
18. White P. Heart disease. 2nd ed. NY: The Macmillan Company; 1938: 128–129.
19. Surawicz B., Fisch C. Cardiac alternans: diverse mechanisms and clinical manifestations. J. Am. Coll. Cardiol. 1992; 20 (2): 483–499. DOI: 10.1016/0735-1097(92)90122-4
20. Spodick D.H. Electric alternation of the heart. Its relation to the kinetics and physiology of the heart during cardiac tamponade. Am. J. Cardiol. 1962; 10: 155–165. DOI: 10.1016/
0002-9149(62)90290-4
21. Brembilla-Perrot B., Lucron H., Schwalm F., Haouzi A. Mechanism of QRS electrical alternans. Heart. 1997; 77 (2): 180–182. DOI: 10.1136/hrt.77.2.180
22. Schulze-Bahr E., Zoelch K.A., Eckardt L., Haverkamp W., Breithardt G., Borggrefe M. Electrical alternans in long QT syndrome resembling a Brugada syndrome pattern. Pacing Clin. Electrophysiol. 2003; 26 (10): 2033–2035. DOI: 10.1046/j.1460-9592.2003.00313.x
23. Chinushi M., Hosaka Y., Washizuka T., Furushima H., Aizawa Y. Arrhythmogenesis of T wave alternans associated with surface QRS complex alternans and the role of ventricular prematurity: observations from a canine model of LQT3 syndrome. J. Cardiovasc. Electrophysiol. 2002; 13 (6): 599–604. DOI: 10.1046/j.1540-8167.2002. 00599.x
24. Moss A.J., Zareba W., Benhorin J., Locati E.H., Hall W.J., Robinson J.L. et al. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation. 1995; 92 (10): 2929–2934. DOI: 10.1161/01.cir. 92.10.2929
Выпуск

Другие статьи выпуска

Цель исследования – изучить структурно-функциональное состояние правых отделов сердца у молодых пациентов с желудочковой электрокардиостимуляцией в отдаленном послеоперационном периоде.
Материал и методы. Обследованы 58 пациентов (34 мужчины и 24 женщины) с имплантированными электрокардиостимуляторами (ЭКС) по поводу атриовентрикулярной (АВ) блокады. В зависимости от причины возникновения АВ-блокады пациентов разделили на две группы. В 1-ю группу (ЭКС+, ВПС+) вошли 28 человек с постоянным ЭКС, имплантированным после хирургической коррекции врожденного порока сердца по поводу возникшей послеоперационной АВ-блокады; во 2-ю группу (ЭКС+, ВПС-) – 30 человек с нехирургической АВ-блокадой, потребовавшей имплантации постоянного ЭКС. Всем пациентам проведены общеклиническое обследование, эхокардиография. Возраст пациентов на момент исследования составил: в 1-й группе – 21,7 (19,2; 23,3) года, во 2-й группе – 22,7 (20,1; 24,7) года; длительность электрокардиостимуляции – 15,9 (13,5; 18,2) и 15,7 (13,9; 18,5) года соответственно. Всем пациентам обеих групп на момент исследования были имплантированы двухкамерные ЭКС со 100% желудочковой стимуляцией.
Результаты. При анализе данных эхокардиографии установлено, что у пациентов 1-й группы статистически значимо большие размеры правого предсердия (ПП) по сравнению со 2-й группой: индекс объема ПП – 27,2 (23,6; 32,2) и 24,2 (21,6; 27,7) мл/м2 соответственно (U = 287,0, р = 0,039). У пациентов 1-й группы в сравнении со 2-й группой установлено значимое снижение показателей продольной функции правого желудочка (ПЖ): значения S’ составили 9,0 (8,3; 10,0) и 11,0 (11,0; 13,0) см/с соответственно (р = 0,000); TAPSE – 15,0 (13,0; 16,0) и 19,0 (18,0; 21,0) мм соответственно (р = 0,000). Показатель фракционного изменения площади (ФИП) в группах значимо не отличался и составил 51,2 (45,2; 56,2) и 46,7 (43,4; 53,2)% соответственно (U = 313,5, р = 0,098). Признаки диастолической дисфункции выявлены у 89% пациентов 1-й группы и 53% исследуемых 2-й группы (F = 0,156, p = 0,004). При оценке тяжести патологии клапанного аппарата правых отделов сердца установлено, что у пациентов 1-й группы выраженная регургитация на трикуспидальном клапане (ТК) имелась в 21%, а умеренно выраженная регургитация на клапане легочной артерии (КЛА) – в 36% случаев. У пациентов 2-й группы выраженная регургитация на ТК не регистрировалась (F = 0,124, p = 0,009) и умеренно выраженная недостаточность на КЛА выявлена у 1 пациента (F = 0,170, p = 0,002). Прогрессирование недостаточности на ТК до умеренной и выше отмечено у 78,5% исследуемых 1-й группы и 94,0% пациентов 2-й группы.
Заключение. У пациентов с послеоперационной АВ-блокадой, возникшей после хирургического лечения ВПС, в отдаленном послеоперационном периоде отмечены расширение полости ПП и снижение продольной функции ПЖ, при нормальных значениях ФИП. У пациентов с нехирургической АВ-блокадой не выявлено значимых нарушений геометрии правых отделов сердца и систолической функции ПЖ при наличии умеренной регургитации на ТК.

Цель исследования – определить представленность повышенной трабекулярности и некомпактной кардиомиопатии в когорте больных с катехоламинергической полиморфной желудочковой тахикардией (КПЖТ).
Материал и методы. В исследование включены 68 больных с КПЖТ, наблюдающихся в Институте им. Ю. Е. Вельтищева. Для выявления наличия структурных изменений всем больным проведена эхокардиография экспертного класса, магнитно-резонансная томография сердца по показаниям.
Результаты. У детей с КПЖТ частота повышенной трабекулярности миокарда левого желудочка / некомпактной кардиомиопатии составила 24% случаев.
Заключение. У больных с КПЖТ ассоциированным структурным изменением миокарда является повышенная трабекулярность миокарда левого желудочка и некомпактная кардиомиопатия.

Цель исследования – проанализировать динамику клинических проявлений у детей с рестриктивной кардиомиопатией (РКМП), обусловленной мутациями в гене TNNI3.
Материал и методы. Проведено исследование, включающее наблюдение за 16 детьми в период с 2013 по 2024 г.
Результаты. У большинства пациентов с генетически детерминированной РКМП, обусловленной мутациями в гене TNNI3, семейный анамнез не был отягощен (94%), характер de novo установлен в 6 (37,5%) исследованных случаях (10 семей не были обследованы из-за отказа родителей). Как минимум 62,5% пациентов достигли конечной точки (летальный исход или трансплантация сердца) за период динамического наблюдения. Отмечалось более тяжелое течение заболевания у детей с дебютом в раннем возрасте на фоне крайне ограниченных возможностей медикаментозной терапии и потребностей в хирургическом лечении в короткие сроки после дебюта заболевания. В качестве иллюстрации приведен клинический пример течения заболевания у ребенка раннего возраста.
Заключение. Генетическая верификация диагноза помогает сделать прогноз заболевания, который является крайне неблагоприятным у пациентов с РКМП, обусловленной мутациями в гене TNNI3, а также решить вопрос о своевременном проведении трансплантации сердца.

Цель исследования – сравнение особенностей течения и исходов саркомерной и РАС-ассоциированной гипертрофической кардиомиопатии (ГКМП) у детей с дебютом на 1-м году жизни.
Материал и методы. По результатам генетического исследования отобраны 36 пациентов с мутациями в генах саркомерных белков и RAS-MAPK сигнального пути. В данных группах проведен сравнительный анализ клинического статуса, лабораторно-инструментальных данных, а также исходов. Разница между группами оценивалась с помощью методов биомедицинской статистики.
Результаты. У 17 (47%) пациентов наблюдались мутации в генах саркомерных белков, у 19 (53%) – в RAS-MAPK сигнального пути. Среди саркомерных мутаций в данной возрастной группе преобладали мутации в гене MYH7 (58,8%, n = 10), а в группе РАС-ассоциированной ГКМП – PTPN11 (42,11%, n = 8) и RAF1 (42,11%, n = 8). У детей с РАСопатиями наблюдалась высокая частота встречаемости врожденных пороков развития органов и систем, а также ВПС (p < 0,05). Бивентрикулярная форма ГКМП выявлена у 14 (38,9%) детей, обструктивная форма – у 17 (47,2%) пациентов. Общая выживаемость на сегодняшний момент (средняя длительность наблюдения 10 [6,0; 20,0] лет) составляет 83,3% (n = 30). Зарегистрировано 6 летальных исходов (по 3 в каждой группе). Общая выживаемость к 5 годам жизни среди всех детей составила 91,7% [95% ДИ 96,4–87,1%], к 10 годам жизни – 88,6% [95% ДИ 93,9–83,2%]. Имплантируемый кардиовертер-дефибриллятор был установлен 4 (23,5%) детям только с саркомерной ГКМП. Миоэктомия проведена 9 (25%) пациентам.
Заключение. Течение ГКМП у детей с дебютом до года характеризуется высокой летальностью. Дети с РАСопатиями представляют особую группу риска в раннем возрасте.

Цель исследования – оценить распространенность тахииндуцированной кардиомиопатии (ТКМП) и выявить факторы, ассоциированные с риском развития ТКМП у детей с идиопатическими желудочковыми аритмиями (ЖА).
Материал и методы. В ретроспективное исследование включены 392 ребенка с идиопатическими ЖА, которым было проведено лечение в НМИЦ им. В. А. Алмазова в 2011–2023 гг. В 1-ю группу вошли 35 (8,9%) пациентов с наличием ТКМП, в группу 2 – 357 (91,1%) пациентов без критериев ТКМП. Всем пациентам с ТКМП проводилась оценка эхокардиографических данных через 2 и 6 мес после старта терапии или проведения катетерной аблации.
Результаты. Распространенность ТКПМ у детей с идиопатическими ЖА составила 8,9%. В ходе исследования были выявлены статистически значимые факторы риска формирования ТКМП у детей с ЖА: возраст 12 лет и старше (p = 0,008), наличие жалоб на повышенную утомляемость (p = 0,040) и пресинкопальные/синкопальные состояния (p = 0,035), наличие парных желудочковых экстрасистол (ЖЭ) (p = 0,010) и желудочковой тахикардии (ЖТ) (p = 0,002). Методом ROC-анализа установлено, что суточная плотность желудочковой эктопии 36,8% и более ассоциируется с развитием ТКМП (p < 0,001). Чувствительность и специфичность модели составили 76,5 и 78,1% соответственно.
Заключение. Такие факторы, как возраст 12 лет и старше, наличие симптомов, парных ЖЭ, ЖТ, а также плотность ЖА, могут использоваться в клинической практике в качестве предикторов формирования ТКМП у детей с идиопатическими ЖА.

Цель исследования – изучить влияние мутаций генов, ответственных за развитие семейной гиперхолестеринемии (СГХС), и липидного профиля на структурно-морфологическое состояние сосудов по показателям толщины комплекса интима–медиа (тКИМ) общей сонной артерии у детей с гетерозиготной семейной гиперхолестеринемией.
Материал и методы. Исследование проводилось в период с 2019 по 2023 г. и включало 214 детей в возрасте от 2 до 17 лет. Группу сравнения составили 107 детей. В основную группу вошли 107 пациентов с диагнозом «семейная гиперхолестеринемия, гетерозиготная форма», которые были разделены на три группы в зависимости от генофенотипа заболевания: группа 2 – дети с положительным фенотипом и с идентифицированными мутациями, ассоциированными с СГХС: LDLR, APOB, PCSK9, LDLRAP1; группа 3 – дети с положительным фенотипом, но не выявленными мутациями; группа 4 – дети с отрицательным фенотипом и положительным генотипом. Всем детям проводились клинико-лабораторная диагностика, УЗИ сосудов шеи с оценкой тКИМ общей сонной артерии. Пациентам основной группы было проведено секвенирование нового поколения с применением панели генов: LDLR, APOB, PCSK9, LDLRAP1.
Результаты. У пациентов 2-й и 3-й групп выявлено статистически значимое увеличение показателей общего холестерина (ОХ), липопротеинов низкой плотности (ЛПНП) и тКИМ относительно группы сравнения. Значения тКИМ были достоверно больше у детей 2-й группы относительно 3-й, при этом показатели липидов у них не отличались. В 4-й группе показатели ЛПНП и тКИМ были достоверно ниже аналогичных показателей 2-й группы. Наиболее распространенными вариантами LDLR оказались: c.986G>A, c.906C>G, c.1187-10G>A. Увеличение тКИМ было достоверно выше у пациентов с c.1187-10G>A относительно c.906C>G.
Заключение. Пациенты с СГХС характеризовались увеличенными уровнями ОХ, ЛПНП и большей тКИМ в отличие от здоровых сверстников. У детей с идентифицированной мутацией в генах, ассоциированных с СГХС, установлены признаки сосудистого ремоделирования с 8-летнего возраста по сравнению с детьми с фенотипической СГХС. Полученные результаты подчеркивают важность ранней диагностики, в том числе генетической, и постоянного мониторинга состояния сосудистой стенки пациентов с СГХС для предотвращения прогрессирования атеросклероза начиная с детского возраста.

Мышечная дистрофия Дюшенна (МДД) в настоящее время представляет собой одну из самых изучаемых прогрессирующих мышечных дистрофий, в отношении которой ведется интенсивная разработка новых фармакологических методов лечения, направленных на коррекцию генетического дефекта и восстановление экспрессии дистрофина. Самыми многообещающими и наиболее перспективными подходами для терапии являются пропуск экзона и технология первичного редактирования с помощью CRISPR/Cas9. Однако, несмотря на эффективность новых методик лечения, большую роль в определении прогноза и продолжительности жизни больных с МДД продолжает играть неуклонно развивающаяся кардиомиопатия, приводящая к декомпенсированной сердечной недостаточности и жизнеугрожающим нарушениям ритма. Прогрессирование кардиомиопатии влияет на доступность всех новых методов лечения, снижая их эффективность и общую функциональную пользу. Поэтому в лечении МДД важен комбинированный подход, включающий в себя как генную терапию, направленную на восстановление экспрессии дистрофина, так и терапию, ориентированную на улучшение выживаемости структурно неполноценных кардиомиоцитов, уменьшение выраженности фиброза миокарда и смягчение дистрофических процессов. В данном обзоре приводится современный взгляд на концепцию единства медикаментозных, хирургических и генотерапевтических методов лечения МДД, целью которых является уменьшение выраженности миокардиальной дисфункции.
Статистика статьи
Статистика просмотров за 2025 год.
Издательство
- Издательство
- ФГБУ НМИЦ ССХ ИМ. А.Н. БАКУЛЕВА МИНЗДРАВА РОССИИ
- Регион
- Россия, Москва
- Почтовый адрес
- 121552, Москва, Рублевское шоссе, д. 135.
- Юр. адрес
- 119049, г Москва, р-н Якиманка, Ленинский пр-кт, д 8
- ФИО
- Голухова Елена Зеликовна (ДИРЕКТОР)
- Контактный телефон
- +7 (495) 4147984
- Сайт
- https://bakulev.ru/